Table of content
Introduction
rboAnalyzer is a tool complementing the BLAST algorithm when searching for a query sequence that is RNA with a secondary structure (which does not have to be known).
The high-scoring pairs (HSPs) in BLAST output are often incomplete (ie. the alignment in HSP does not cover the whole query sequence). This is a major drawback when trying to characterize the potential ncRNA indicated by the HSP.
Therefore, rboAnalyzer tries to find full-length RNA sequences from the incomplete HSPs from the BLAST output and predict their secondary structures with one or more methods. Score for similarity (as proxy to homology) between the estimated full-length sequence and query sequence is also computed.
This webserver is used for interactive analysis of individual high scoring pairs (HSPs) from NCBI BLAST webserver output, for bulk analysis or results obtained from custom database see the commandline application.
Searching sequence database with BLAST
Since BLAST scoring parameters can significantly influence the number and length of obtained HSPs we suggest to test out multiple BLAST parameter groups to find out which provides relevant results for your use-case. Analysis with rboAnalyzer is helpful by providing potential secondary structure and genomic context information to the HSPs of interest, especially for HSPs with partial alignment or alignment with many mismatches and/or gaps. Additionally it is possible to explore and combine multiple BLAST parameter settings in addition to the default ones, as no single ones are appropriate for all situations (ref. 1).
Here we list scoring parameters that where used for ncRNA searches (ref. 2-4) and recommended for cross-species exploration and RNA search (ref. 5):
| Parameter | ref. 2 | ref. 3 | ref. 4 | ref. 5 |
|---|---|---|---|---|
| MatchReward | 5 | 5 | 5 | 1 |
| MismatchPenalty | -4 | -4 | -4 | -1 |
| GapOpeningCost | 10 | 8 | 10 | 1 |
| GapExtensionCost | 10 | 6 | 6 | 2 |
| WordSize | 7 | 4 | 7 | 9 |
Functionality overview

The rboAnalyzer has 3 stages:
- Extension of full-length RNA sequence from HSP.
- Estimation of homology of full-length matches to query sequence.
- Prediction of secondary structures.
Methods for extending the partial matches to full length
We offer 3 methods for extending the partial matches.
simple
This means that location of extended full-length sequence is computed from lengths of unaligned parts of query sequence on 5’ and 3’ ends of the HSP. (fast)
locarna
In this method, the loci at the subject sequence containing partial match with flanking regions is realigned to the query sequence with Locarna algorithm. The sequence aligned to the query is considered to be the extended full-length sequence.
meta
Here the two aforementioned methods are combined and extended full-length sequences are scored with covariance model. The better scoring sequence is chosen. (most accurate)
simple

In the simple mode we compute the location of the extended subject sequence according to the unaligned parts of the query sequence, i.e. those that were not aligned in HSP and flank the HSP alignment at both 5’ and 3’ ends. In this toy example we have the Plus/Plus BLAST HSP with a section of the query sequence between nucleotides 10 and 21 aligned to a section of the subject sequence between nucleotides 1000 and 1009. The task is to extend the partial HSP subject sequence between nucleotides 1000 and 1009 to the length of the query sequence.
Suppose that the query sequence (the red bar in the figure of the example) is 50 bases long. Then the length of the unaligned part of the query at 5’ end is 9 nucleotides (subtract nucleotide positions 10 - 1) and the length of the unaligned part of the query at 3’ end is 29 nucleotides (subtract positions 50 - 21). The positions of the extended subject sequence at the whole subject sequence is computed by adding/subtracting the lengths of the unaligned parts of the query sequence to/from 3’/5’ ends of the partial HSP subject sequence, respectively. Then, 5’ and 3’ ends of the extended HSP sequence lie at nucleotides 991 (1000 – 9) and 1038 (1009 + 29), respectively. Because there are 2 gaps in HPS subject sequence, the resulting extended subject sequence will be 2 nucleotides shorter than the query sequence.
Theoretically, the 2 extra nucleotides could be added at 3’ end of the extended subject sequence to make it as long as the query sequence. But this might not be biologically relevant as the gap could occur naturally instead of being caused by the alignment.
locarna

With locarna mode we first extract so called supersequence, which is region on subject sequence as with simple, extended on 5’ and 3’ ends by extra regions from the subject sequence. This supersequence is then realigned with Locarna algorithm to obtain the extended partial match.
The Locarna algorithm utilises possible pairings in it’s computations, thus it is better suited to align RNAs then BLAST algorithm. The Locarna is by default called with free-endgaps=++++ parameters. Additionally the information about matching nucleotides from BLAST HSPs is used to construct so called anchor for the Locarna algorithm. The anchor defines columns of alignment which are considered aligned. As the anchor we consider consecutive series of matches of length at least L in BLAST HSP. The default value of L is 7.
This way the alignment is anchored and the Locarna algorithm can align query to the supersequence. With the free-endgaps=++++ option, the algorithm does not put penalty to unaligned ends of supersequence. The estimated full-length sequence is the continuous part of supersequence aligned to the query sequence.
To set custom options for locarna please use the rboAnalyzer pipeline.
meta

This approach combines the simple and locarna extension methods. It extends the HSP with both methods and then selects the extended sequence with with higher score to the covariance model. This is repeated for each HSP to be extended. The different color of the sequences in the diagram illustrates that the extended sequences are slightly different.
Estimation of homology
Here we compute score for relation between the extended full-length sequence and query sequence. The computation is based on aligning covariance model (CM) to extended full-length sequence with cmalign program from the Infernal package.
We offer 3 options on how to provide covariance model:
build with RSEARCH (default)
By default we build the covariance model from the query sequence (secondary structure predicted by RNAfold) and RIBOSUM65 matrix.
infer from Rfam
The Rfam database is searched for the best reliable matching model with the query sequence. From the found covariance models (if any) the one with highest score and lowest p-value is selected, if these conditions are not satisfied by single model, the search is not considered reliable and no model is reported as matched. The reliably matched model is used for homology inference.
supply your own model
If you have the covariance model, you pass it directly to the system. The provided covariance model, will also be used for predicting the secondary structures with relevant prediction methods.
Secondary structure prediction
Following prediction methods were selected from commandline rboAnalyzer based on their suitability for fast interactive prediction of individual secondary structures from extended partial matches.
RNAfold
de novo secondary structure prediction from extended partial match with RNAfold program, part of ViennaRNA Package.

cm-Rc
Align covariance model (either best matching Rfam CM or user supplied CM) to extended partial match with cmalign program, use refold.pl to extract conserved base-pairs and use them as constraints for RNAfold prediction.
This method may not be available, if no covariance model was provided or if none were reliably found in Rfam database.

cm-sub
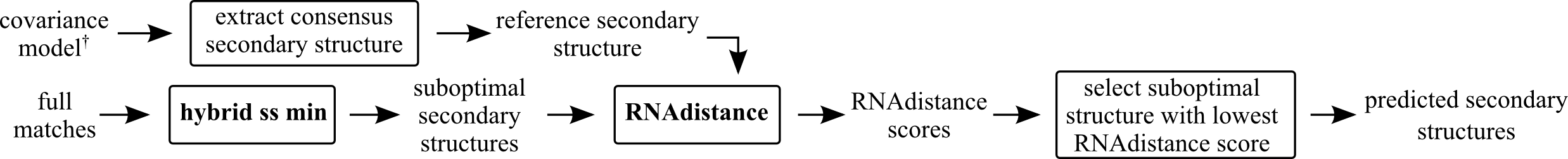
Extract consensus secondary structure from covariance model (either best matching Rfam CM or user supplied CM), predict suboptimal secondary structures with hybrid-ss-min and select the most similar suboptimal secondary structure from the predicted ones (lowest RNAdistance score).
This method may not be available, if no covariance model was provided and none were reliably found in Rfam database.
Parameters hybrid-ss-min man page (part of UnaFold):
- mfold percent (
P) indicates the percent suboptimality to consider, only structures with energies within P% of the minimum will be output - mfold window (
W) indicates the window size; a structure must have at least W basepairs that are each a distance of at least W away from any basepair in a previous structure - mfold max (
X) represents an absolute limit on the number of structures computed
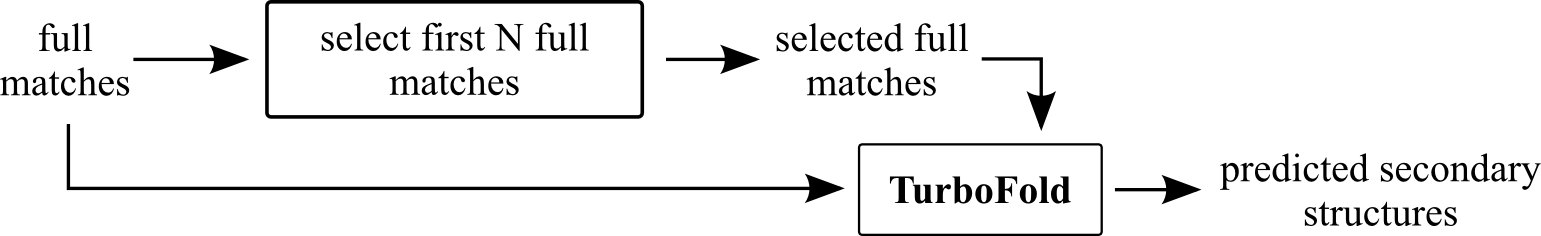
Turbo-fast
This method is based on TurboFold software (RNAstructure package).
First create group of reference sequences (non-redundant) starting with query and adding first N-1 extended sequences that have bit-score > 0, their length differ at maximum from query length of QlenMax and are different from sequences already in he group.
For each extended full match, we make new non-redundant group of sequences consisting of the extended full match for which we want to predict secondary structure and up to N-1 reference sequences.
This method will not be used if the extended sequence contains ambiguous bases.
Parameters:
- N - required number of sequences in prediction group
- Query Max len diff - Maximum allowed difference of extended full match from query sequence (fraction)
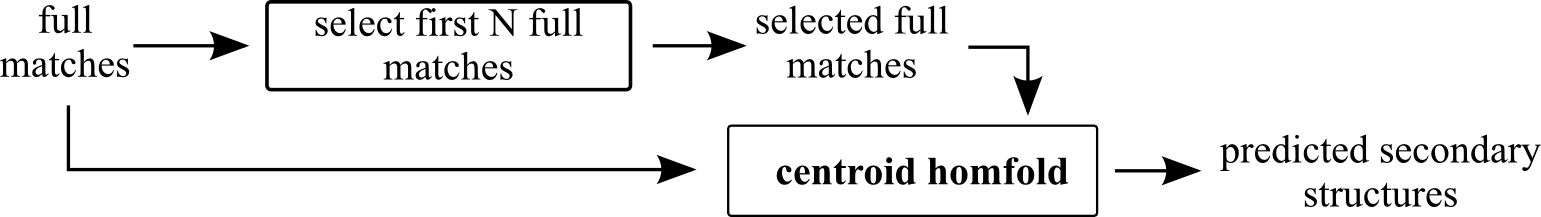
centroid-fast
First create group of reference sequences (non-redundant) starting with query and adding first N-1 extended sequences that have bit-score > 0, their length differ at maximum from query length of QlenMax and are different from sequences already in he group.
Use this group as the homologous sequence group for prediction with centroid homfold.
Parameters:
- N - required number of sequences in prediction group
- Query Max len diff - Maximum allowed difference of extended full match from query sequence (fraction)
centroid homfold parameter: - BP inference engine - see centroid homfold documentation
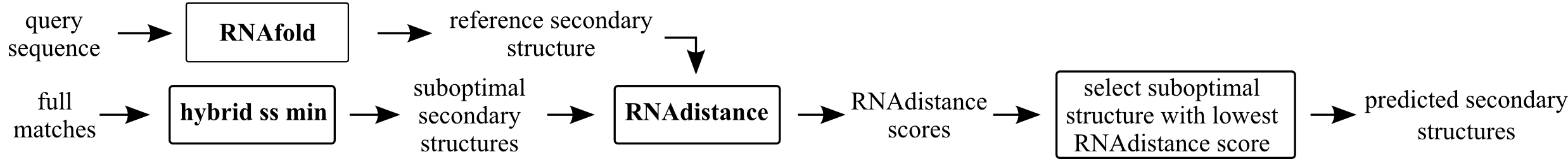
fq-sub
Predict secondary structure for query sequence with RNAfold. Then for the extended full match predict suboptimal secondary structures and select the most similar one to the secondary structure predicted for query sequence (lowest RNAdistance score).
Parameters hybrid-ss-min man page (part of UnaFold):
- mfold percent (
P) indicates the percent suboptimality to consider, only structures with energies within P% of the minimum will be output - mfold window (
W) indicates the window size; a structure must have at least W basepairs that are each a distance of at least W away from any basepair in a previous structure - mfold max (
X) represents an absolute limit on the number of structures computed
Usage
Prerequisites
A modern browser with javascript enabled is required to use this webserver. The site was tested with Mozilla Firefox and Google Chrome. Please use one of these.
Input form
For the analysis to work we need the query sequence used for searching the nucleotide database and the BLAST output.
For every input field there are basic format checks in place. Possible problems will be reported and must be fixed before job submission.
![]()
Query sequence
Query sequence must be in FASTA format, e.g.:
>my_query_sequence CGATGCATGCAGCTGTACGTAGCTGTCGATThe query sequence can’t contain ambiguous bases (only
ACTGare accepted)Only one query sequence is allowed
You can type in the query sequence or upload file containing the query sequence
BLAST output
- We accept BLAST output in two formats, either
XML(preferred) ortext - The
textoutput must be the “classic” format (output format 0, can be retrieved fromTraditional Results page) - Only BLAST output for one query sequence at a time is supported.
- All sequences must be available from NCBI ENTREZ. This should be without any issue when using NCBI’s nucleotide databases. The
Accession.Versionuniquely identifies the sequence. TheAccessionis and alphanumerical sequence identifier while theVersionis a number which identifies the sequence revision (starting with 1). More information about theAccession.Versionidentifiers can be found here and here. - You can type in, or upload the data. If your search is recent, then you can type in the NCBI’s
RIDand pull the BLAST output directly to the input form. - Most sequences present in NCBI’s BLAST databases are preloaded on the server, if they are not available, they are downloaded as part of the HSP analysis.
- Note that the sequences with nonstandard accession numbers that start with GPS_ are obtained from NCBI without annotations and only when needed.
Using RID for BLAST output
To retrieve the BLAST output directly from NCBI you need valid RID (e.g. K4S3S11T01R - these are stored at NCBI only for limited time). Type in the RID to NCBI BLAST RID input and click on Retrieve RID button in the Input form section. The server will attempt to obtain the provided RID from NCBI. If successful, the obtained data will appear as xml text in the gray area.
Extension mode
You can choose from simple, locarna and meta extension methods. The default simple is chosen for speed.
For locarna and meta you can alter extension parameters (for simple there are no parameters).
- Extra area around extended hit - length of extra region on 5’ and 3’ end
- Locarna anchor length - minimal number of consecutive matches in BLAST HSP to define an anchor for locarna

Covariance model
Choose the source of covariance model for estimating homology.
- Infer from data (description)
- Infer from Rfam (description)
- User supplied - Supply your own covariance model. It will be used for both - homology inference and secondary structure prediction. Only one model per file is allowed.

Methods for secondary structure prediction
None or all prediction methods can be selected by clicking the sliders on the left of the prediction method name. Three methods are selected by default (RNAfold, cm-Rc, Turbo-fast).
Methods allowing to set parameters will show the parameters input if selected.
Available prediction methods:
- RNAfold
- cm-Rc
- Turbo-fast
- cm-sub
- centroid-fast
- fq-sub

Job submission
If all checks are OK you can submit the job to the server (otherwise the Submit button will be disabled). the Additional checks on the input data will be performed.

If everything is in order the Input section will collapse and the Matched CM and Results section will appear.
Your session will get assigned unique identifier (UUID) which will appear in the address bar (after the “?q=”). Copy or bookmark this address to maintain access to your results. The UUID is the sole identifier for your results and the only way to access them.
No cookies are stored by our server in your browser.
Matched CM section
Upon job submission the server tries to find most similar reliable covariance model for the query sequence in Rfam database. If model is found it is reported here.
This section also reports the RNAdistance score for predicting secondary structure for query sequence with cm-Rc to covariance model consensus structure. This score serves as reference for the reported RNAdistance scores for the predicted secondary structures of the full matches.
Results
The results present interactive web page, where you can analyze HSPs of interest by clicking the Analyze button for the HSP.
Each HSP from the input has it’s own panel. Initially only the HSP alignment is shown and the analysis is triggered by clicking the Analyze button.
Results of the analysis are loaded as they are completed.
- Progress in each stage of computation is reported and upon completion the result is automatically loaded
- In case that the HSP is for the sequence not available in server’s database, the sequence will be retrieved from NCBI.
- If TurboFold is selected as one of the methods for secondary structure prediction, sufficient number of extended sequences must be obtained for prediction and the prediction of secondary structure for individual HSPs will be blocked until then.
Filters
The HSPs can be filtered with Filters dropdown (any active filters are indicated by blue color).
- by BLAST E-value: display only HSPs within specified range
- by BLAST score: display only HSPs within specified range
- title contains: display only HSPs whose title or accession number contains specified text
- by status: applies only to already analyzed HSPs, filters by automatically determined category.
You can export extended sequences or secondary structures by clicking the copy icon at the HSP, or by selecting one or multiple HSPs (checkbox on the right) and using the Export dropdown.
You also can export offline copy of the results (using the Export dropdown), with all the computed data present (however, no new HPSs can be extended and interactive genome browser is replaced with picture of the genome loci).
The fasta-like format containing secondary structures
>uid:N|ACCESSION.VERSION|STRAND|START:END DESCRIPTION
SEQUENCE
SECONDARY_STRUCTURE_1 PREDICTION_METHOD_1
SECONDARY_STRUCTURE_2 PREDICTION_METHOD_2
# - where the N is serial number of BLAST HSP
# - ACCESSION.VERSION are the sequence identifiers
# - STRAND is sequence strand in relation to database record
# - START:END format where START is always lower index then END (direction is defined by "direction")
# - DESCRIPTION database sequence description
# the sequence is always given 5' to 3' direction
# Example
>uid:3|LR535856.1|-|20229035:20229107 Mastacembelus armatus genome assembly, chromosome: 24
GCCUCAUAGCUCAGAGGUUUAGAGCACUGGUCUUGUAAACCAGGGGUCGUGAGUUCGAGUCUCACUGGGGCCU
(((((...((((.........)))).(((((.......))))).....(((((.......))))).))))).. Turbo-fast
((((((..((((.........)))).(((((.......))))).....(((((.......))))))))))).. cm-Rc
((((((.(((((.(((.((..((.(.(((((.......))))).).))..)).)))))).))...)))))).. rnafold
Examples
The input form contains several example inputs and one pre-computed example output.
The example inputs contains query sequences and BLAST outputs for following RNAs
To paste the example input, click on the RNA initials in the “Input” header. On click the query sequence and BLAST output are inserted in corresponding boxes. You can choose the extension mode, covariance model source and the prediction methods that will be used. Then continue by clicking on the “Submit” button.
The precomputed output example is for MS1 RNA and can be loaded here.
Step by step example - tRNA
The tRNA from Escherichia coli was searched for in NCBI nt database limited to fishes.
To input the query sequence and BLAST output click on the tRNA button in Examples section.

Then select locarna in extension mode and leave default parameters.

Proceed with submitting the input form (the Submit button might be disabled if problems are detected in the form).
The server will process the input and Matched CM and Results section will appear.
- The
Matched CMpresents results of Rfam family identification for query RNA and the RNAdistance score for secondary structure of the query sequence predicted by cm-Rc method using tRNA model obtained from Rfam database. - The
Resultssection display individual HSPs which can be individually analyzed.
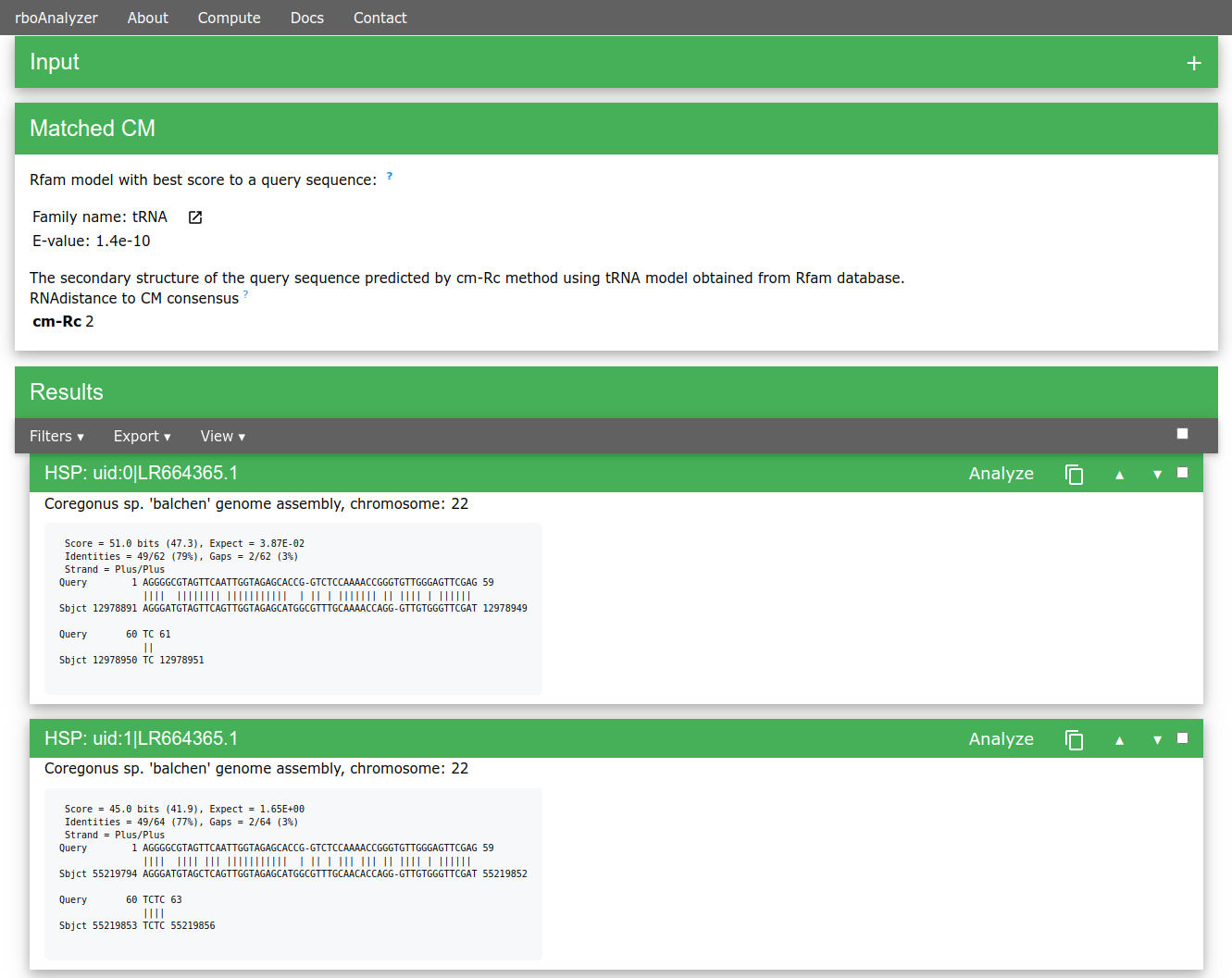
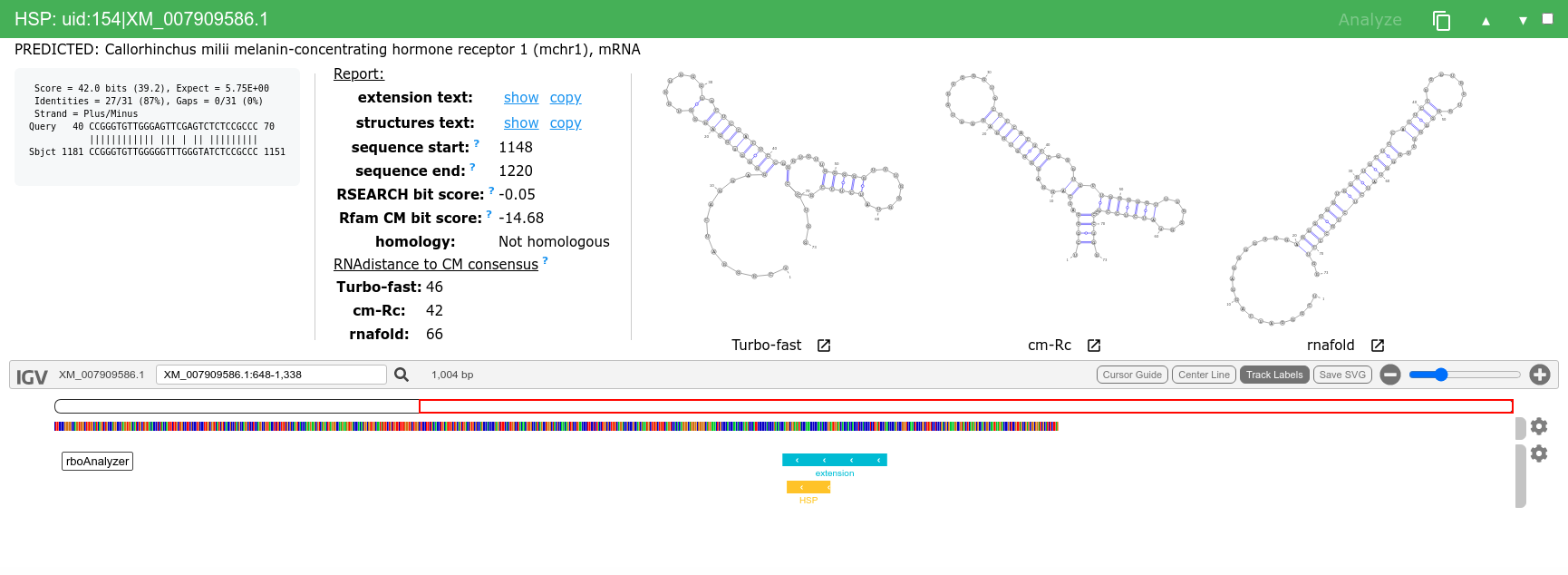
We can then analyze and inspect HSPs of interest e.g. uid:3|LR535856.1, uid:7|LR664378.1, uid:146|AP008946.1 or uid:154|XM_007909586.1.
Extended sequence for uid:3 HSP has 74 nt (from 44 nts of the HSP), the RNAdistance from Rfam consensus the cm-Rc predicted structure is only 3 which is almost precise match.
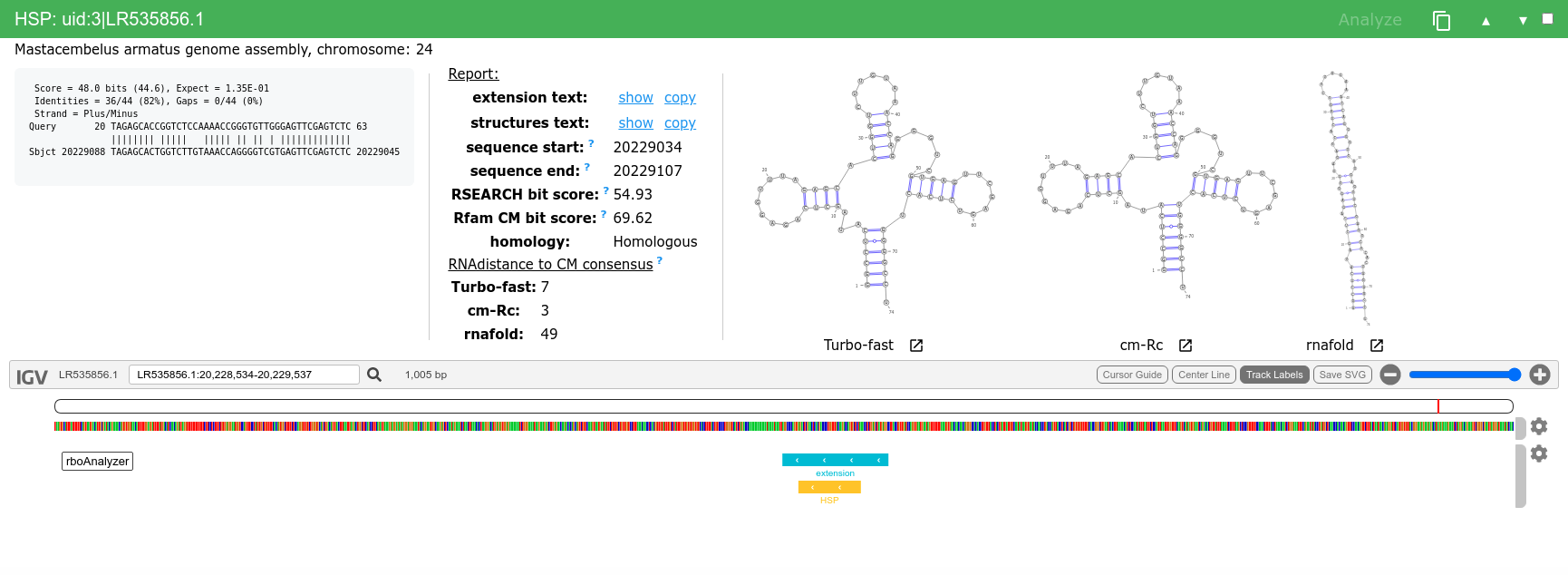
Extended uid:7 HSP is flagged by Uncertain homology (based on RSEARCH bit score) and you can see that even the predicted secondary structures are not similar to the Rfam consensus structure, even though that the BLAST score of this HSP is 47 and that of the uid is comparable 48.
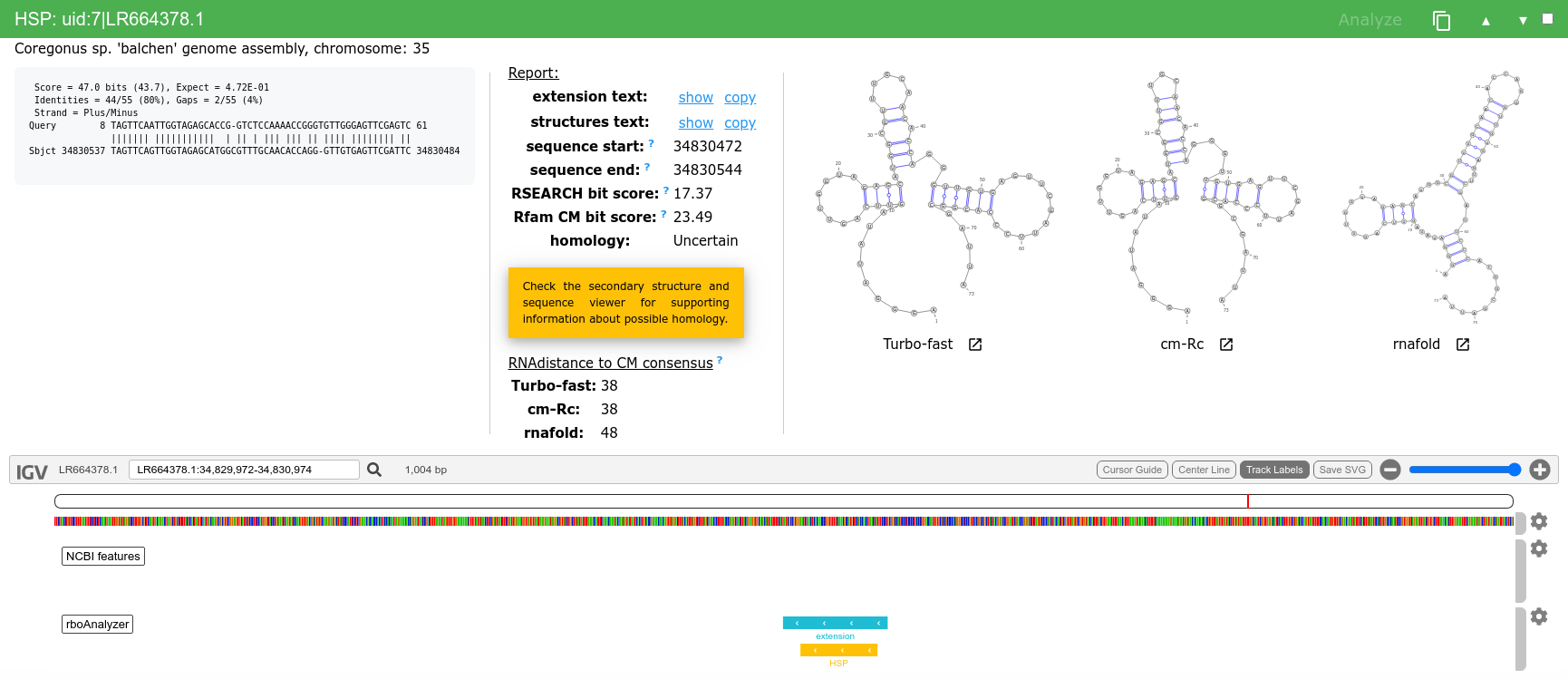
This HSP represents example of very short, otherwise likely discarded, HSP which can be extended to full length sequence with very similar predicted secondary structures.
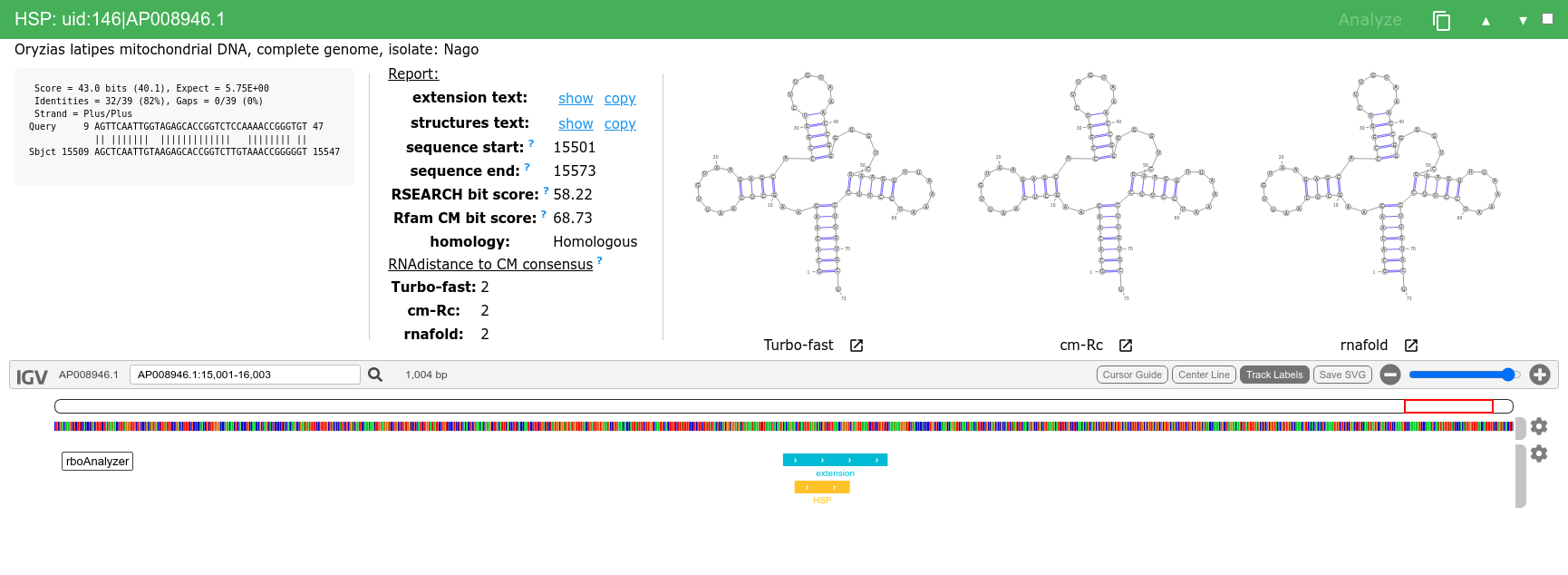
Finally, when the extended sequence is dissimilar to the query sequence it is reflected in the RSEARCH (Rfam CM) score and in the predicted secondary structures.
References
The webserver uses following resources:
- Rfam - covariance models from Rfam database - Ioanna Kalvari, Eric P Nawrocki, Nancy Ontiveros-Palacios, Joanna Argasinska, Kevin Lamkiewicz, Manja Marz, Sam Griffiths-Jones, Claire Toffano-Nioche, Daniel Gautheret, Zasha Weinberg, Elena Rivas, Sean R Eddy, Robert D Finn, Alex Bateman, Anton I Petrov, Rfam 14: expanded coverage of metagenomic, viral and microRNA families, Nucleic Acids Research, Volume 49, Issue D1, 8 January 2021, Pages D192–D200, https://doi.org/10.1093/nar/gkaa1047
- NCBI - nucleotide sequences from NCBI’s repository - NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018 Jan 4;46(D1):D8-D13. doi: 10.1093/nar/gkx1095. PMID: 29140470; PMCID: PMC5753372.
The webserver is build with and uses following software:
- Elm - functional language for web frontend
- igv.js - embeddable genome browser - preprint
- bioperl - Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, Lapp H, Lehväslaiho H, Matsalla C, Mungall CJ, Osborne BI, Pocock MR, Schattner P, Senger M, Stein LD, Stupka E, Wilkinson MD, and Birney E. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002 Oct;12(10):1611-8.
- NCBI C++ toolkit - The NCBI C++ Toolkit (https://ncbi.github.io/cxx-toolkit/) by the National Center for Biotechnology Information, U.S. National Library of Medicine; Bethesda MD, 20894 USA.
- SQLite
- PostgreSQL
- VARNA - RNA visualization applet - Kévin Darty, Alain Denise, Yann Ponty, VARNA: Interactive drawing and editing of the RNA secondary structure, Bioinformatics, Volume 25, Issue 15, 1 August 2009, Pages 1974–1975, https://doi.org/10.1093/bioinformatics/btp250
- RNA image scaling is based on elm-2d-viewer
- UNAFold - UNAFold: Markham N.R., Zuker M. (2008) UNAFold. In: Keith J.M. (eds) Bioinformatics. Methods in Molecular Biology™, vol 453. Humana Press. https://doi.org/10.1007/978-1-60327-429-6_1
The webserver builds on rboAnalyzer pipeline, we list it’s dependencies also here for clarity.
- ViennaRNA: Lorenz, Ronny and Bernhart, Stephan H. and Höner zu Siederdissen, Christian and Tafer, Hakim and Flamm, Christoph and Stadler, Peter F. and Hofacker, Ivo L. ViennaRNA Package 2.0. https://doi.org/10.1186/1748-7188-6-26.
- Locarna: Sebastian Will, Tejal Joshi, Ivo L. Hofacker, Peter F. Stadler, and Rolf Backofen. LocARNA-P: Accurate boundary prediction and improved detection of structural RNAs RNA, 18 no. 5, pp. 900-14, 2012. https://doi.org/10.1261/rna.029041.111, website
- NCBI BLAST: Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., & Madden T.L. (2008) “BLAST+: architecture and applications.” BMC Bioinformatics 10:421. https://doi.org/10.1186/1471-2105-10-421, website
- RSEARCH: Finding Homologs of Single Structured RNA Sequences. R. J. Klein, S. R. Eddy. BMC Bioinformatics, 4:44, 2003. https://doi.org/10.1186/1471-2105-4-44, website
- Infernal: Infernal 1.1: 100-fold Faster RNA Homology Searches. E. P. Nawrocki, S. R. Eddy. Bioinformatics, 29:2933-2935, 2013. https://doi.org/10.1093/bioinformatics/btt509, website
- Clustal omega: Sievers F, Wilm A, Dineen DG, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7:539. https://doi.org/10.1038/msb.2011.75, website
- MUSCLE: Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput Nucleic Acids Res. 32(5):1792-1797. https://doi.org/10.1093/nar/gkh340, website
- Centroid homfold: Michiaki Hamada, Koichiro Yamada, Kengo Sato, Martin C. Frith, Kiyoshi Asai; CentroidHomfold-LAST: accurate prediction of RNA secondary structure using automatically collected homologous sequences, Nucleic Acids Research, Volume 39, Issue suppl_2, 1 July 2011, Pages W100–W106. https://doi.org/10.1093/nar/gkr290, github
- TurboFold (RNAstructure): Tan, Z., Fu, Y., Sharma, G., & Mathews, D. H. (2017). TurboFold II: RNA structural alignment and secondary structure prediction informed by multiple homologs. Nucleic Acids Research. 45: 11570-11581. https://doi.org/10.1093/nar/gkx815, website
- Biopython: Cock, P.J.A. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009 Jun 1; 25(11) 1422-3. http://dx.doi.org/10.1093/bioinformatics/btp163, website
- NumPy
- Pandas
- Jinja2
- Matplotlib
References concerning BLAST parameters:
- Velandia-Huerto, C. A., Gittenberger, A. A., Brown, F. D., Stadler, P. F., & Bermúdez-Santana, C. I. (2016). Automated detection of ncRNAs in the draft genome sequence of a colonial tunicate: the carpet sea squirt Didemnum vexillum. BMC genomics, 17(1), 1-15.
- Freyhult, E. K., Bollback, J. P., & Gardner, P. P. (2007). Exploring genomic dark matter: a critical assessment of the performance of homology search methods on noncoding RNA. Genome research, 17(1), 117-125.
- Roshan, U., Chikkagoudar, S., & Livesay, D. R. (2008). Searching for evolutionary distant RNA homologs within genomic sequences using partition function posterior probabilities. BMC bioinformatics, 9(1), 1-9.
- Mount, S., & Nguyen, M.-C. (2006, December 14). BLASTN parameters for noncoding queries. Retrieved May 16, 2022, from http://stevemount.outfoxing.com/Posting0004.html
- Korf, I., Yandell, M., & Bedell, J. (2003). Blast (1st ed.). O’Reilly Media.
Changelog
25/10/2024
- added Rfam 15.0; 15.0 is now the default Rfam version.
24/11/2023
- added Rfam 14.9 and 14.10; 14.10 now default Rfam version.
1/6/2022
- added Rfam 14.8, now default Rfam version.
19/5/2022
- added paragraph about BLAST searches to the documentation
7/1/2022
- added Rfam 14.7, now default option.
6/10/2021
- fix offline igv rendering issue (introduced by undocumented behaviour of 2.9.1 version)
30/9/2021
- Allow users to select Rfam version.
3/9/2021
- Rfam updated to 14.6
21/7/2021
- Rfam updated to 14.5
- igv.js updated to 2.9.1, switching to CSI index for gff files
19/1/2021
- Rfam updated to 14.4
Funding
This work was supported by ELIXIR CZ research infrastructure project (MEYS Grant No: LM2015047) including access to computing and storage facilities.

This work was supported from European Regional Development Fund - Project “ELIXIR-CZ: Budování kapacit” (No. CZ.02.1.01/0.0/0.0/16_013/0001777).

rboAnalyzer version 0.1.5a1